publications
publications by categories in reversed chronological order. generated by jekyll-scholar.
Journal Articles
2026
- JEP
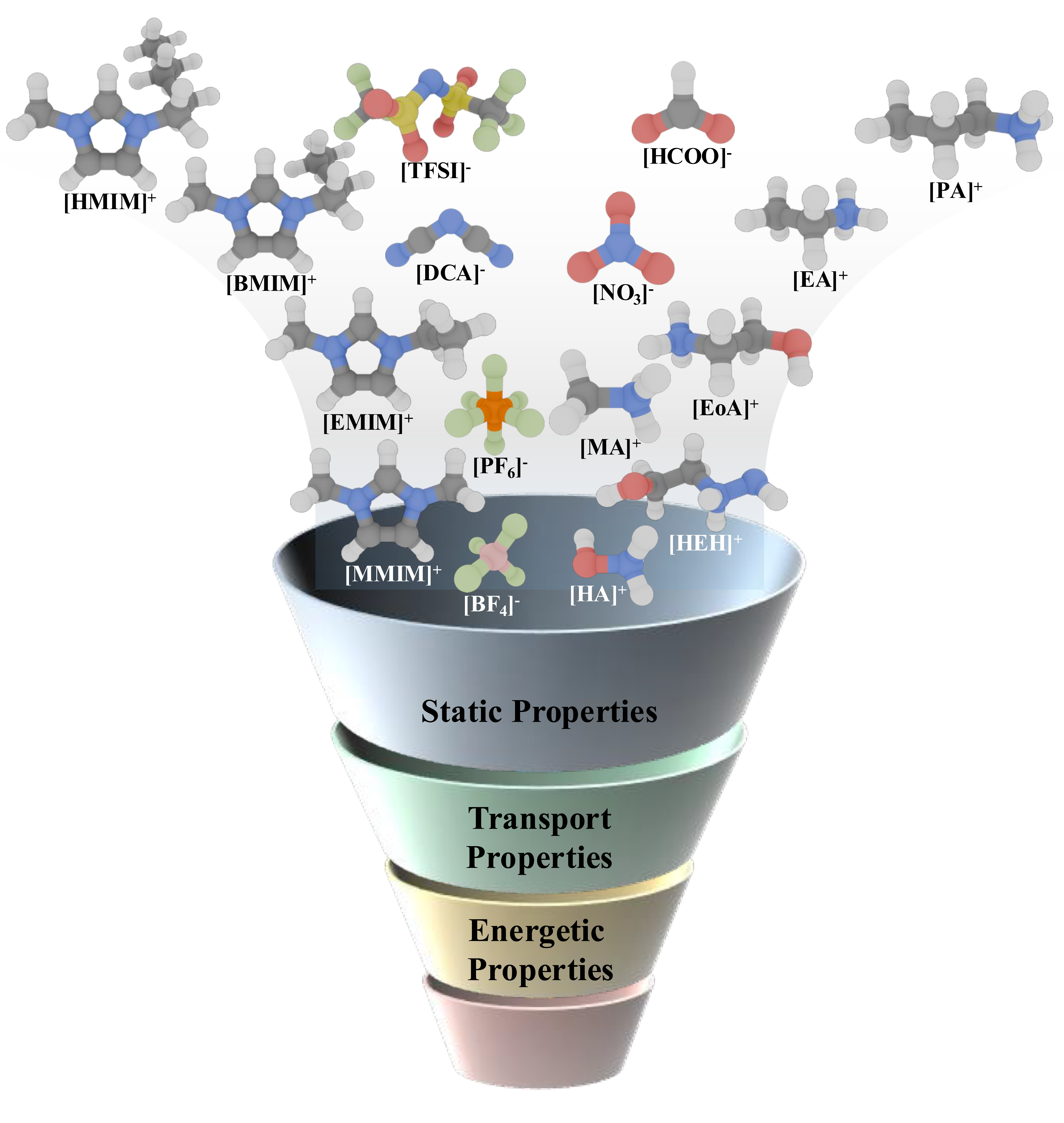 Multimode propellant discovery: a computational high-throughput screening paradigmShehan M. Parmar, Orion Cohen, Kristin A. Persson, and 3 more authors2026
Multimode propellant discovery: a computational high-throughput screening paradigmShehan M. Parmar, Orion Cohen, Kristin A. Persson, and 3 more authors2026The emergence of multimode propulsion (MMP) architectures for in-space propulsion shows great promise for improving the future of spacecraft mission design. However, seeking ideal propellants requires traversing a complex chemical design space that satisfies optimal chemical and electric propulsion requirements. To this end, we propose a computational, high-throughput screening framework founded upon software developed by the Materials Project and accurate, polarizable molecular dynamics (MD) force fields to discover new MMP propellants. We highlight the metric space within the scope of this framework and present a case study of a candidate ionic liquid (IL) propellant mixture composed of hydroxylammonium nitrate (HAN) and hydroxyethylhydrazinium nitrate (HEHN) to inform important paths for the future.
@article{parmar2026_multimode, author = {Parmar, Shehan M. and Cohen, Orion and Persson, Kristin A. and Vaghjiani, Ghanshyam L. and McDaniel, Jesse G. and Wirz, Richard E.}, title = {Multimode propellant discovery: a computational high-throughput screening paradigm}, journal = {Journal of Electric Propulsion}, volume = {5}, number = {1}, doi = {10.1007/s44205-025-00174-6}, year = {2026}, type = {journal} }

2025
- Digit. Discov.
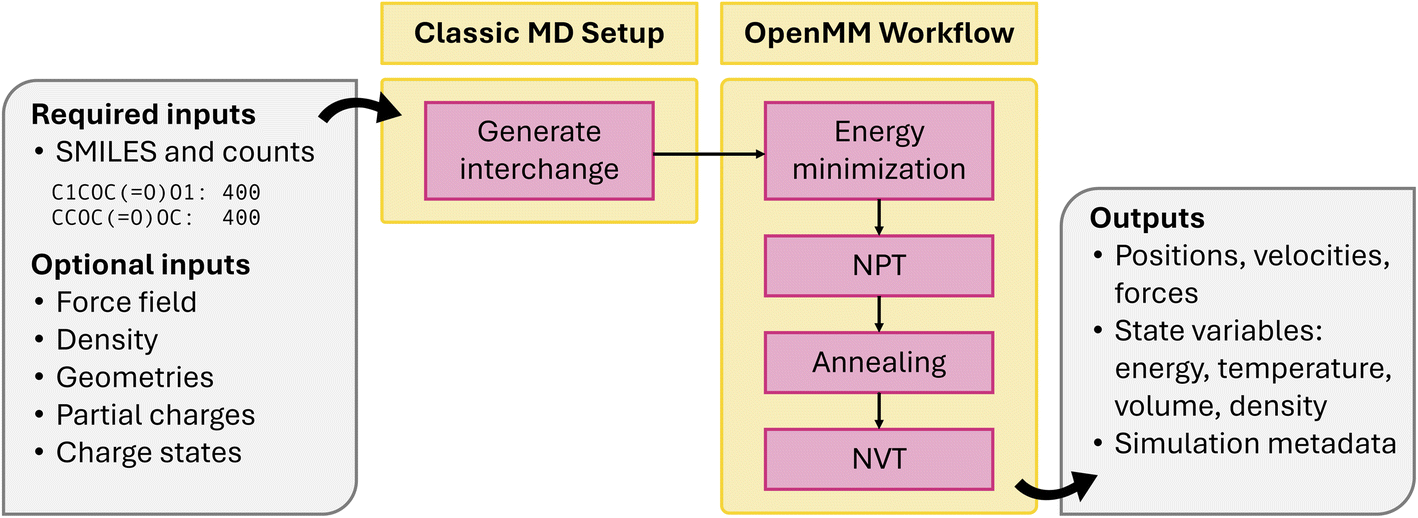 Atomate2: modular workflows for materials scienceAlex M. Ganose, Hrushikesh Sahasrabuddhe, Mark Asta, and 53 more authors2025
Atomate2: modular workflows for materials scienceAlex M. Ganose, Hrushikesh Sahasrabuddhe, Mark Asta, and 53 more authors2025High-throughput density functional theory (DFT) calculations have become a vital element of computational materials science, enabling materials screening, property database generation, and training of "universal" machine learning models. While several software frameworks have emerged to support these computational efforts, new developments such as machine learned force fields have increased demands for more flexible and programmable workflow solutions. This manuscript introduces atomate2, a comprehensive evolution of our original atomate framework, designed to address existing limitations in computational materials research infrastructure. Key features include the support for multiple electronic structure packages and interoperability between them, along with generalizable workflows that can be written in an abstract form irrespective of the DFT package or machine learning force field used within them. Our hope is that atomate2’s improved usability and extensibility can reduce technical barriers for high-throughput research workflows and facilitate the rapid adoption of emerging methods in computational material science.
@article{ganose2025_atomate2, author = {Ganose, Alex M. and Sahasrabuddhe, Hrushikesh and Asta, Mark and Beck, Kevin and Biswas, Tathagata and Bonkowski, Alexander and Bustamante, Joana and Chen, Xin and Chiang, Yuan and Chrzan, Daryl C. and Clary, Jacob and Cohen, Orion A. and Ertural, Christina and Gallant, Max C. and George, Janine and Gerits, Sophie and Goodall, Rhys E. A. and Guha, Rishabh D. and Hautier, Geoffroy and Horton, Matthew and Inizan, T. J. and Kaplan, Aaron D. and Kingsbury, Ryan S. and Kuner, Matthew C. and Li, Bryant and Linn, Xavier and McDermott, Matthew J. and Mohanakrishnan, Rohith Srinivaas and Naik, Aakash N. and Neaton, Jeffrey B. and Parmar, Shehan M. and Persson, Kristin A. and Petretto, Guido and Purcell, Thomas A. R. and Ricci, Francesco and Rich, Benjamin and Riebesell, Janosh and Rignanese, Gian-Marco and Rosen, Andrew S. and Scheffler, Matthias and Schmidt, Jonathan and Shen, Jimmy-Xuan and Sobolev, Andrei and Sundararaman, Ravishankar and Tezak, Cooper and Trinquet, Victor and Varley, Joel B. and Vigil-Fowler, Derek and Wang, Duo and Waroquiers, David and Wen, Mingjian and Yang, Han and Zheng, Hui and Zheng, Jiongzhi and Zhu, Zhuoying and Jain, Anubhav}, title = {Atomate2: modular workflows for materials science}, journal = {Digital Discovery}, year = {2025}, doi = {10.1039/D5DD00019J}, google_scholar_id = {W7OEmFMy1HYC}, type = {journal} }

2024
- JPC B
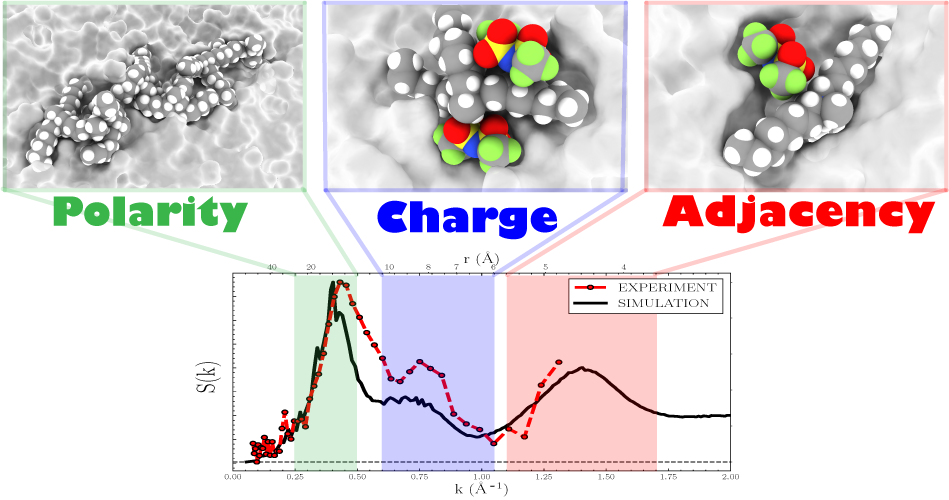 Structural Properties of [N1888][TFSI] Ionic Liquid: A Small Angle Neutron Scattering and Polarizable Molecular Dynamics StudyShehan M. Parmar, William Dean, Changwoo Do, and 4 more authors2024
Structural Properties of [N1888][TFSI] Ionic Liquid: A Small Angle Neutron Scattering and Polarizable Molecular Dynamics StudyShehan M. Parmar, William Dean, Changwoo Do, and 4 more authors2024In this study, we investigate the quaternary ammonium-based ionic liquid (QAIL), methyltrioctylammonium bis(trifluoromethylsulfonyl)imide, [N1888][TFSI], utilizing small angle neutron scattering (SANS) measurements and polarizable molecular dynamics (MD) simulations to characterize the short- and long-range liquid structure. Scattering structure factors show signatures of three length scales in reciprocal space indicative of alternating polarity (k ∼ 0.44 Å–1), charge (k ∼ 0.75 Å–1), and neighboring or adjacent (k ∼ 1.46 Å–1) domains. Excellent agreement between simulation and experimental scattering structure factors validates various simulation analyses that provide detailed atomistic characterization of the different length scale correlations. The first solvation shell structure is illustrated by obtaining radial, angular, dihedral, and combined distribution functions, where two dominant spatial motifs, N+···N– and N+···O–, compete for optimal packing around the polar head of the [N1888]+ cation. Intermediate and long-range structures are governed by the balance between local electroneutrality and octyl chain networking, respectively. By computing the charge-correlation structure factor, SZZ, and the spatial extent of the octyl chain network using graph theory, the bulk-phase structure of [N1888][TFSI] is characterized in terms of electrostatic screening and apolar domain formation length scales.
@article{parmar2024_N1888_TFSI, author = {Parmar, Shehan M. and Dean, William and Do, Changwoo and Browning, James F. and Klein, Jeffrey M. and Gurkan, Burcu E. and McDaniel, Jesse G.}, title = {Structural Properties of [N1888][TFSI] Ionic Liquid: A Small Angle Neutron Scattering and Polarizable Molecular Dynamics Study}, journal = {The Journal of Physical Chemistry B}, volume = {128}, number = {45}, pages = {11313-11327}, doi = {10.1021/acs.jpcb.4c06255}, year = {2024}, google_scholar_id = {Y0pCki6q_DkC}, type = {journal} } - JPC B
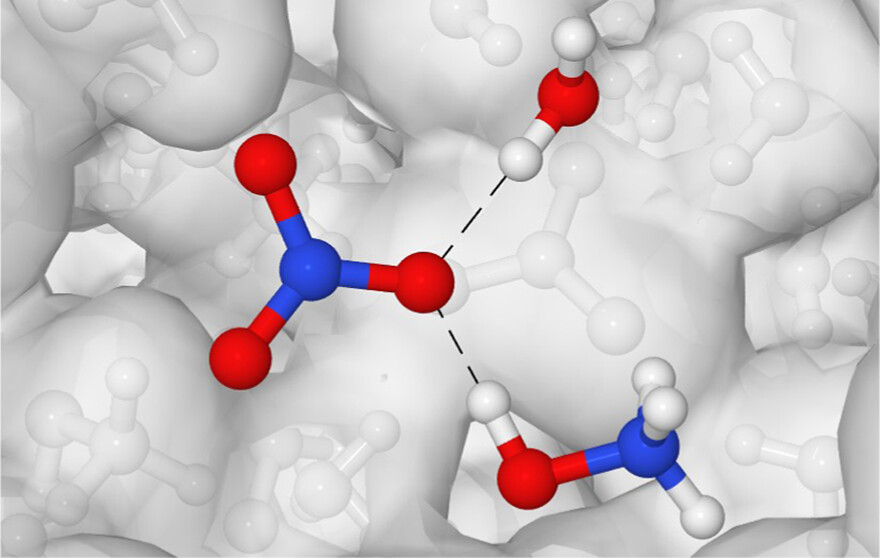 Liquid Structure and Hydrogen Bonding in Aqueous Hydroxylammonium NitrateDaniel D. Depew, Ghanshyam L. Vaghjiani, Shehan M. Parmar, and 1 more author2024
Liquid Structure and Hydrogen Bonding in Aqueous Hydroxylammonium NitrateDaniel D. Depew, Ghanshyam L. Vaghjiani, Shehan M. Parmar, and 1 more author2024Hydroxylammonium nitrate (HAN) has emerged as a promising component in ionic liquid-based spacecraft propellants. However, the physicochemical and structural properties of aqueous HAN have been largely overlooked. The purpose of this study is to investigate the hydrogen bonding in aqueous HAN and understand its implications on these properties and the proton transfer mechanism as a function of concentration. Classical polarizable molecular dynamics simulations have been employed with the APPLE&P force field to analyze the geometry of individual hydrogen bonds and the overall hydrogen-bonding network in various concentrations of aqueous HAN. Radial distribution functions (RDFs) and spatial distribution functions (SDFs) indicate the structural arrangement of the species and their hydrogen bonds. Projections of water density and the orientation of its electric dipole moment near the ions provide insight into the hydrogen-bonding network. The incorporation of water into the hydrogen-bonding network at high ion concentrations occurs via interstitial accommodation around the ions immediately outside the first solvation shell. While ion pairs are observed at all concentrations considered, the frequency of Ha···On hydrogen bonds increases substantially with the ion concentration. The findings contribute to a better fundamental understanding of HAN and the precursors of reactivity, crucial to the development of “green” spacecraft propellants.
@article{depew_parmar2024_HAN_H2O, author = {Depew, Daniel D. and Vaghjiani, Ghanshyam L. and Parmar, Shehan M. and Wang, Joseph J.}, title = {Liquid Structure and Hydrogen Bonding in Aqueous Hydroxylammonium Nitrate}, journal = {The Journal of Physical Chemistry B}, volume = {128}, number = {3}, pages = {824-840}, doi = {10.1021/acs.jpcb.3c05623}, year = {2024}, google_scholar_id = {IjCSPb-OGe4C}, type = {journal} }


2023
- JPC B
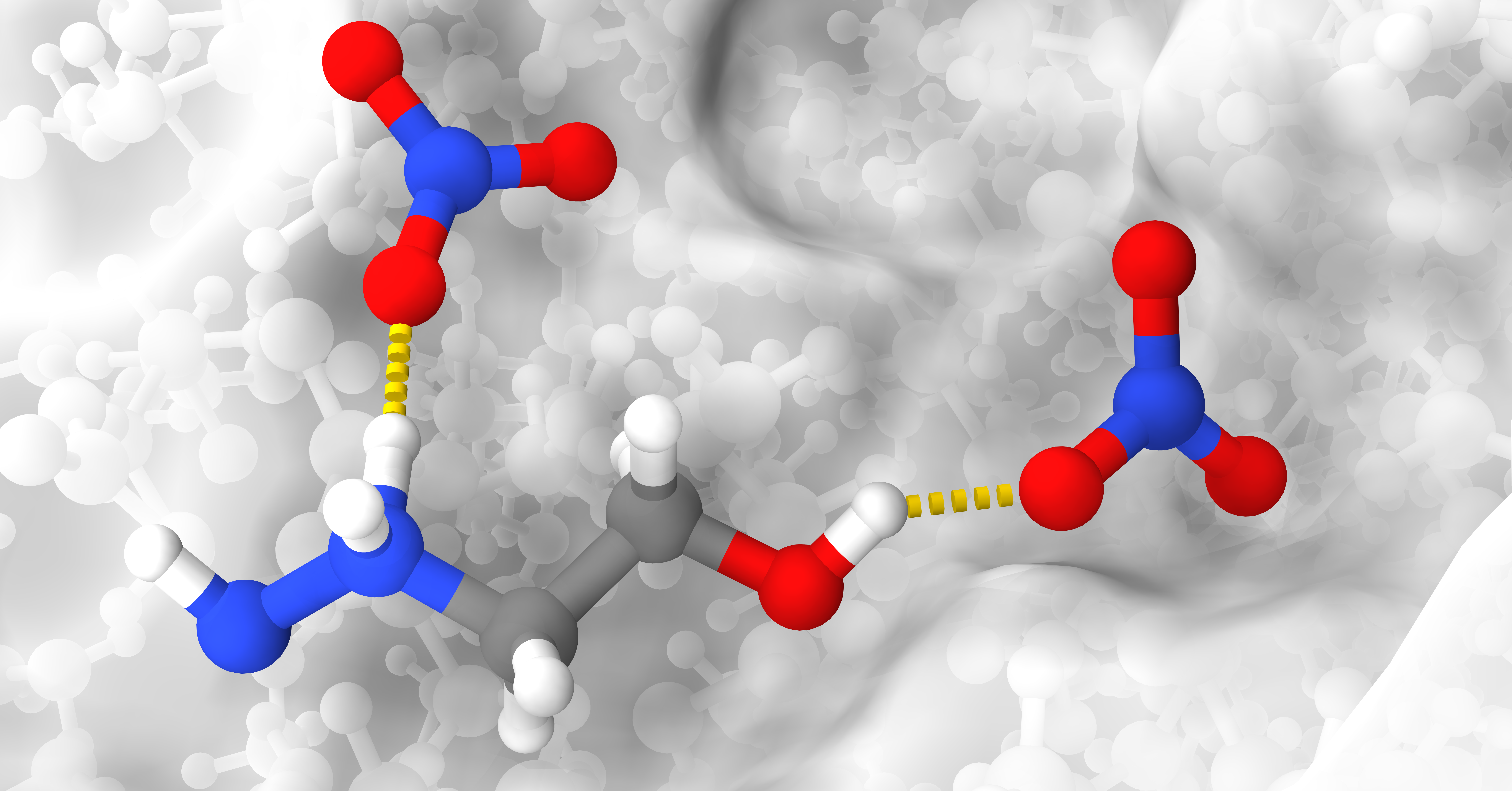 Structural Properties of HEHN- and HAN-Based Ionic Liquid Mixtures: A Polarizable Molecular Dynamics StudyShehan M. Parmar, Daniel D. Depew, Richard E. Wirz, and 1 more author2023
Structural Properties of HEHN- and HAN-Based Ionic Liquid Mixtures: A Polarizable Molecular Dynamics StudyShehan M. Parmar, Daniel D. Depew, Richard E. Wirz, and 1 more author2023ACS Editors’ Choice® is a collection designed to feature scientific articles of broad public interest.
Molecular dynamics simulations of binary mixtures comprising 2-hydroxyethylhydrazinium nitrate (HEHN) and hydroxylammonium nitrate (HAN) were conducted using the polarizable APPLE&P force field to investigate fundamental properties of multimode propulsion (MMP) propellants. Calculated densities as a function of temperature were in good agreement with experiments and similar simulations. The structural properties of neat HEHN and HAN–HEHN provided insights into their inherent, protic nature. Radial distribution functions (RDFs) identified key hydrogen bonding sites located at N–H···O and O–H···O within a first solvation shell of approximately 2 Å. Angular distribution functions further affirmed the relatively strong nature of the hydrogen bonds with nearly linear directionality. The increased hydroxylammonium cation (HA+) mole fraction shows the influence of competitively strong hydrogen bonds on the overall hydrogen bond network. Dominant spatial motifs via three-dimensional distribution functions along with nearly nanosecond-long hydrogen bond lifetimes highlight the local bonding environment that may precede proton transfer reactions.
@article{parmar2023_HAN_HEHN, author = {Parmar, Shehan M. and Depew, Daniel D. and Wirz, Richard E. and Vaghjiani, Ghanshyam L.}, title = {Structural Properties of HEHN- and HAN-Based Ionic Liquid Mixtures: A Polarizable Molecular Dynamics Study}, journal = {The Journal of Physical Chemistry B}, volume = {127}, number = {40}, pages = {8616-8633}, keywords = {ionic liquids, polarizable force fields, molecular dynamics}, doi = {10.1021/acs.jpcb.3c02649}, year = {2023}, google_scholar_id = {UeHWp8X0CEIC}, type = {journal} }

2022
- JEP
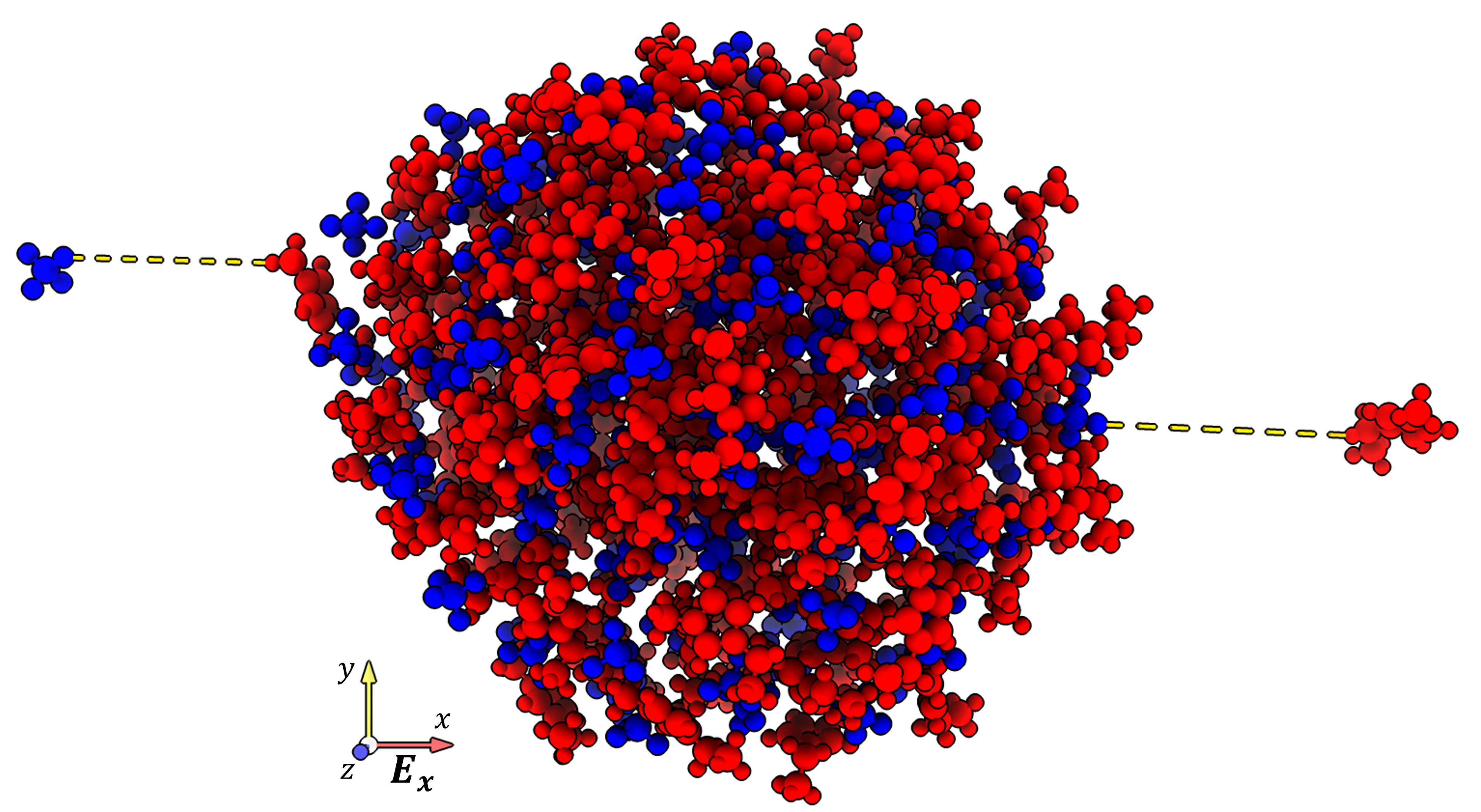 Molecular Dynamics Simulations of Ion Extraction from Nanodroplets for Ionic Liquid Electrospray ThrustersTakaaki Enomoto, Shehan M. Parmar, Ryohei Yamada, and 2 more authors2022
Molecular Dynamics Simulations of Ion Extraction from Nanodroplets for Ionic Liquid Electrospray ThrustersTakaaki Enomoto, Shehan M. Parmar, Ryohei Yamada, and 2 more authors2022Molecular dynamics (MD) simulations were performed for ion extraction from electrospray thrusters to investigate relevant extraction processes numerically. To approximate the electrospray jet tip, a simulation domain consisting of 4-5 nm-sized ionic liquid droplets was used. The extracted ion angles and kinetic energies from EMI–BF4 (1-ethyl-3-methylimidazolium tetrafluoroborate) and EMI–Im (1-ethyl-3-methylimidazolium bis((trifluoromethyl)sulfonyl)imide) droplets were quantified by applying uniform electric fields of 1.3–1.7 V nm−1. The MD simulations are in great agreement with simulations presented in the literature and consistently show a greater preference for monomer emission than reported experimentally. At field strengths above 1.5 V nm−1, apparent droplet fracturing and breakup lead to an increase in ion angular velocity distributions. Greater mobility of EMI–BF4 ions than EMI–Im was also observed, indicative of the crucial role of cation-anion hydrogen bond strengths in ion extraction and beam composition between different propellants.
@article{parmar2022_EMIM_EMI-BF4, author = {Enomoto, Takaaki and Parmar, Shehan M. and Yamada, Ryohei and Wirz, Richard E. and Takao, Yoshinori}, title = {Molecular Dynamics Simulations of Ion Extraction from Nanodroplets for Ionic Liquid Electrospray Thrusters}, journal = {Journal of Electric Propulsion}, volume = {1}, number = {13}, doi = {10.1007/s44205-022-00010-1}, year = {2022}, google_scholar_id = {2osOgNQ5qMEC}, type = {journal} }

Conference Proceedings
2024
- IEPC
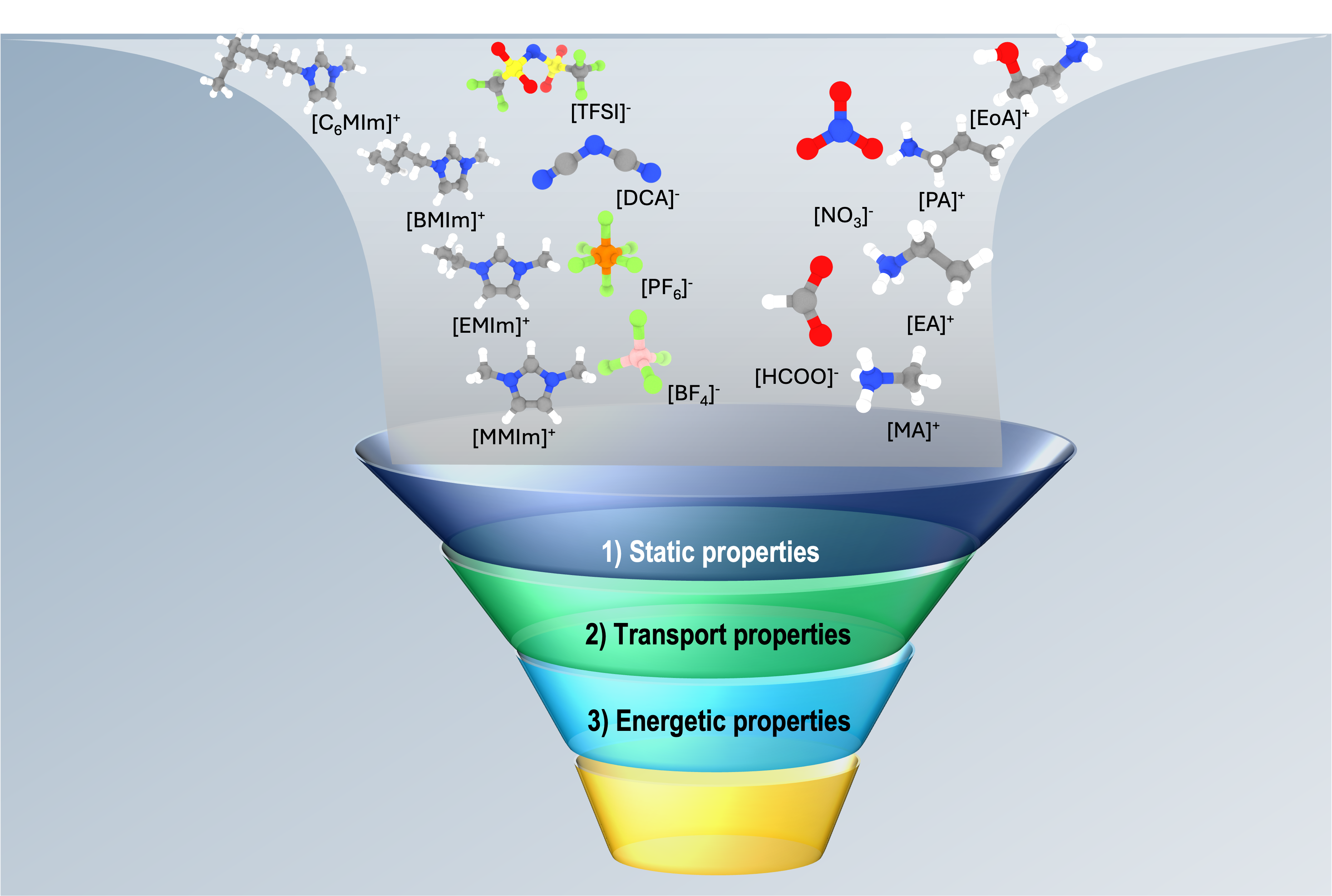 Multimode Propellant Discovery: A Computational High-throughput Screening ParadigmShehan M. Parmar, Orion Cohen, Kristin A. Persson, and 3 more authorsIn International Electric Propulsion Conference, Toulouse, France, Jun 2024
Multimode Propellant Discovery: A Computational High-throughput Screening ParadigmShehan M. Parmar, Orion Cohen, Kristin A. Persson, and 3 more authorsIn International Electric Propulsion Conference, Toulouse, France, Jun 2024Best Paper of the Electrostatic Thrusters Session
The emergence of multimode propulsion (MMP) architectures in in-space propulsion shows great promise for improving the future of spacecraft mission design. However, seeking an ideal propellant requires traversing a complex chemical design space that satisfies optimal chemical and electric propulsion requirements. To this end, we propose a computational, high-throughput screening framework founded upon software developed by the Materials Project and accurate, polarizable molecular dynamics (MD) force fields to discover new MMP propellants. We highlight the metric space within the scope of this framework and present a case study of a candidate ionic liquid propellant mixture composed of HAN and HEHN to inform important paths for the future.
@inproceedings{parmar2024_multimode, author = {Parmar, Shehan M. and Cohen, Orion and Persson, Kristin A. and Vaghjiani, Ghanshyam L. and McDaniel, Jesse G. and Wirz, Richard E.}, title = {Multimode Propellant Discovery: A Computational High-throughput Screening Paradigm}, booktitle = {International Electric Propulsion Conference}, year = {2024}, month = jun, address = {Toulouse, France}, url = {https://www.researchgate.net/profile/Shehan-Parmar/publication/382817540_Multimode_Propellant_Discovery_A_Computational_High-throughput_Screening_Paradigm/links/66ad049251aa0775f264aab5/Multimode-Propellant-Discovery-A-Computational-High-throughput-Screening-Paradigm.pdf}, }

2022
- IEPC
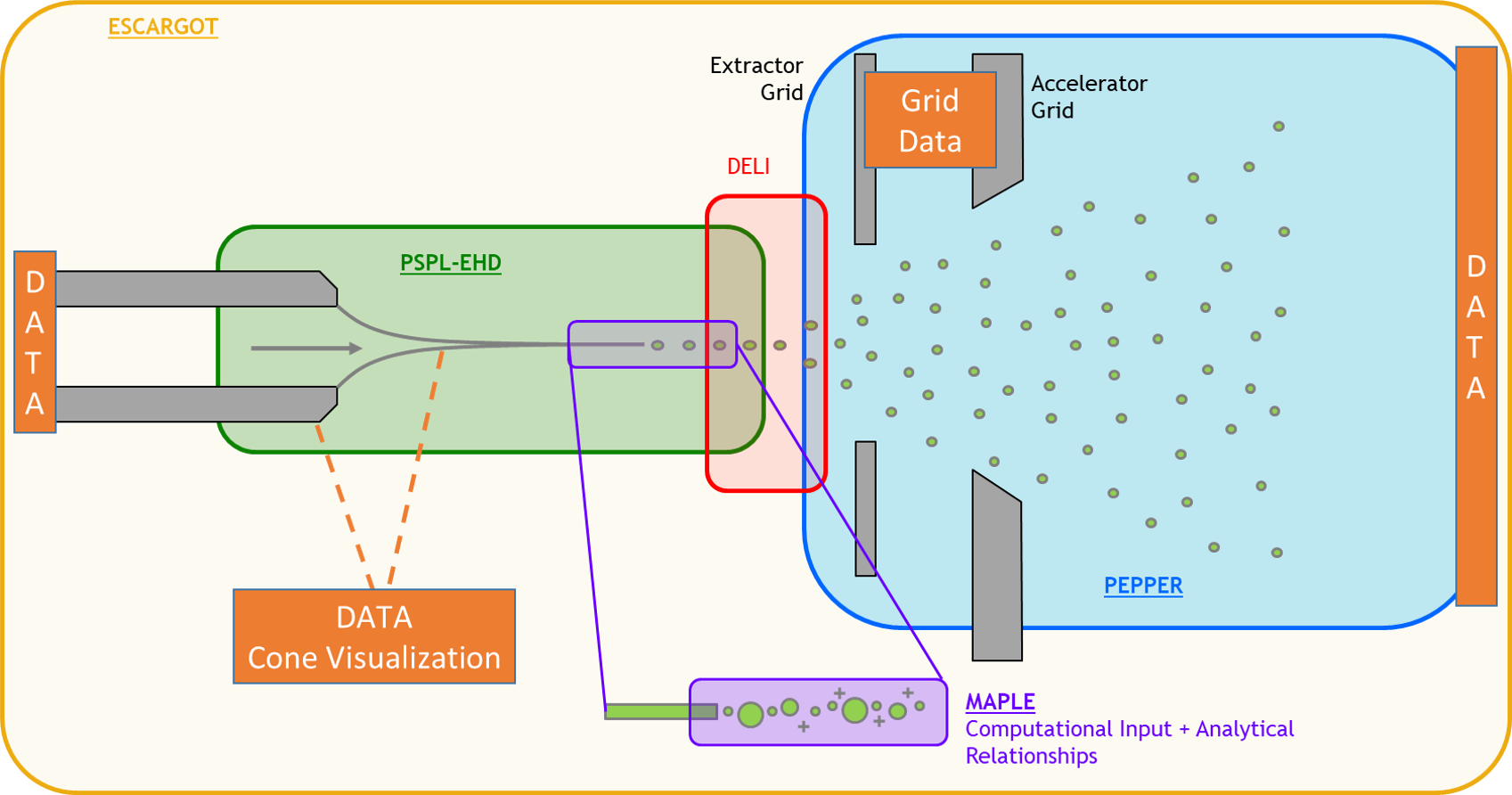 A Bayesian Data-driven Model for Quantifying Electrospray LifetimeShehan M. Parmar, Adam Collins, and Richard E. WirzIn International Electric Propulsion Conference, Boston, MA, Jun 2022
A Bayesian Data-driven Model for Quantifying Electrospray LifetimeShehan M. Parmar, Adam Collins, and Richard E. WirzIn International Electric Propulsion Conference, Boston, MA, Jun 2022To mitigate primary electrospray failure mechanisms and improve overall performance, a complete understanding of plume evolution is necessary. To this end, a data-driven modeling framework has been developed to elucidate emission behavior that leads to experimentally observed mass flux and current density profiles. The relevant computational domain explored in this work assumes plume species motion that is dominated by solely external electric fields generated by extractor and accelerator electrodes. Consequently, charged particles with similar charge-specific kinetic energies starting at the same initial positions follow similar trajectories, thereby reducing the parameter space necessary to fully capture possible ion and droplet trajectories propagated in the Plume Region. The physics-based model was emulated by a surrogate model using a polynomial chaos expansion (PCE) to map model inputs to final line-of-sight (LOS) angles. The PCE was evaluated just as a simple, analytical equation to accelerate computational time within a Bayesian inference framework. The unknown distribution in upstream input conditions for plume species emission angles was quantified with uncertainty bounds, showing the need for super-Gaussian-like distributions similar to downstream mass flux profiles. Initial findings from Gaussian mixture modeling (GMM) show the existence of latent, sub-profiles within a super-Gaussian profile, indicative of dissimilar species created upon emission.
@inproceedings{parmar2022_bayesian, author = {Parmar, Shehan M. and Collins, Adam and Wirz, Richard E.}, title = {A Bayesian Data-driven Model for Quantifying Electrospray Lifetime}, booktitle = {International Electric Propulsion Conference}, year = {2022}, month = jun, address = {Boston, MA}, url = {https://www.researchgate.net/publication/362080112_A_Bayesian_Data-driven_Model_for_Quantifying_Electrospray_Lifetime}, } - AIAA
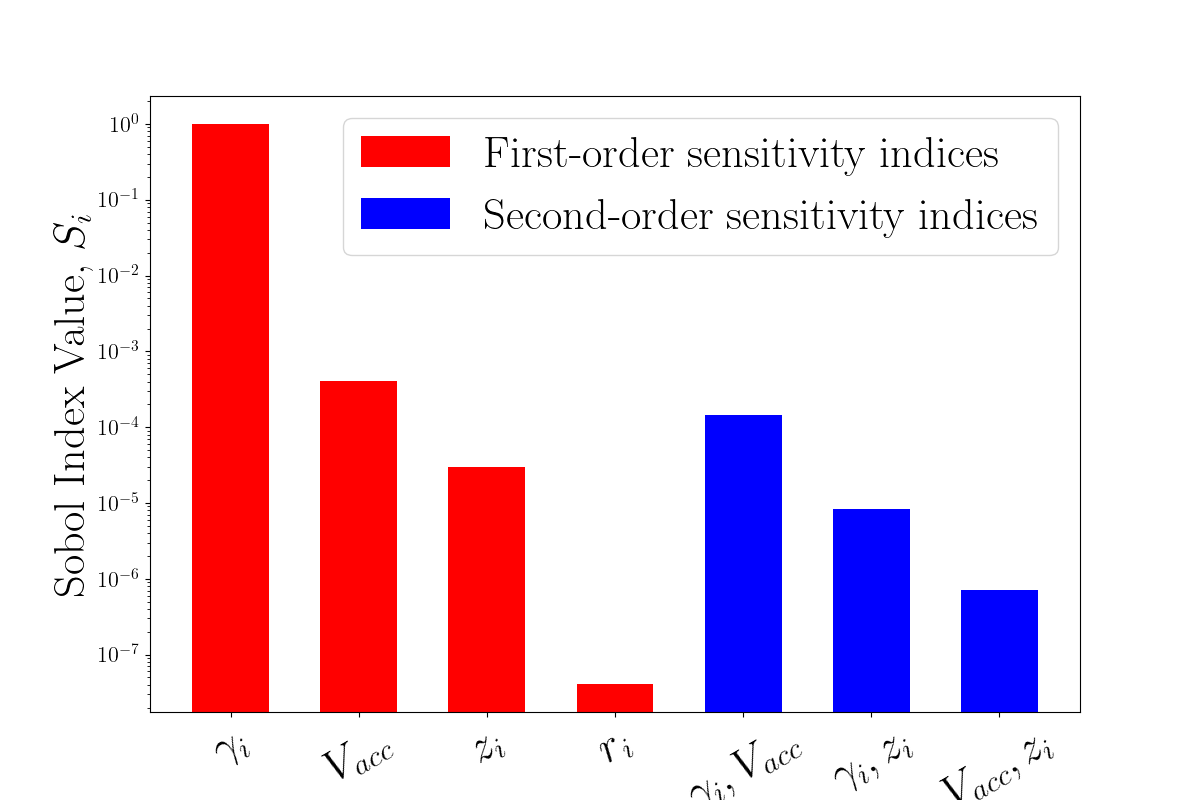 Electrospray Plume Modeling for Rapid Life and Performance AnalysisShehan M. Parmar, Adam Collins, and Richard E. WirzIn AIAA SciTech Forum and Exposition, San Diego, CA, Jan 2022
Electrospray Plume Modeling for Rapid Life and Performance AnalysisShehan M. Parmar, Adam Collins, and Richard E. WirzIn AIAA SciTech Forum and Exposition, San Diego, CA, Jan 2022A data-driven, reduced-order electrospray plume modeling framework has been developed to predict downstream mass flux evolution and thruster lifetime. The computational domain is the Plume Region, where charged plume species motion can be assumed to be governed solely by electrostatic forces. To enable a surrogate modeling approach, physics-based charged particle simulations were reduced to a single, analytical equation using polynomial chaos expansion (PCE) as the functional form. This model was then evaluated within a Bayesian inference framework using electrospray plume mass flux measurements taken at the UCLA Plasma & Space Propulsion Laboratory (PSPL). The unknown distribution in upstream input conditions for plume species emission angles was quantified with uncertainty bounds, showing the need for super-Gaussian-like distributions similar to downstream mass flux profiles. Model-predicted mass flux profiles indicate regions of higher uncertainty at wider angles, indicating the need for further experimental results at these locations.
@inproceedings{parmar2022_plume, author = {Parmar, Shehan M. and Collins, Adam and Wirz, Richard E.}, title = {Electrospray Plume Modeling for Rapid Life and Performance Analysis}, booktitle = {AIAA SciTech Forum and Exposition}, year = {2022}, month = jan, address = {San Diego, CA}, doi = {10.2514/6.2022-1357}, }


2019
- AIAAThermal Decomposition of Hydroxylammonium Nitrate: ReaxFF Training Set Development for Molecular Dynamics SimulationsDaniel Depew, Joseph Wang, Shehan M. Parmar, and 4 more authorsIn AIAA Propulsion and Energy Forum and Exposition, Indianapolis, IN, Aug 2019Technical Paper Session
A ReaxFF reactive force field training set has been developed for the thermal decomposition of hydroxylammonium nitrate (HAN). The training set consists of geometries, partial atomic charges, and energy barriers for a number of reactions relevant to HAN thermal decomposition. Geometries and partial atomic charges were calculated in both the gas phase and in solution at the M06-2X/aug-cc-pVTZ level of theory. The SMD-GIL solvent model was used to approximate a high concentration of HAN in solution. Transition states for elementary reactions were found at the GIL/ωB97X-D/6-311++G** level of theory. An important autocatalytic pathway for the regeneration of HONO in HAN decomposition is discussed. The training set from this work can be used to train a ReaxFF force field capable of conducting reactive molecular dynamics simulations of HAN thermal decomposition.
@inproceedings{depew2019_reaxff, author = {Depew, Daniel and Wang, Joseph and Parmar, Shehan M. and Chambreau, Steven and Bedrov, Dmitry and van Duin, Adri and Vaghjiani, Ghanshyam}, title = {Thermal Decomposition of Hydroxylammonium Nitrate: ReaxFF Training Set Development for Molecular Dynamics Simulations}, booktitle = {AIAA Propulsion and Energy Forum and Exposition}, year = {2019}, month = aug, address = {Indianapolis, IN}, note = {Technical Paper Session}, doi = {10.2514/6.2019-4367}, }